Autoimmune Thyrotoxicosis: Diagnostic Challenges
Abstract
Autoimmune thyrotoxicosis or Graves’ disease (GD) is the most common cause of hyperthyroidism in the United States. GD occurs more often in women (ratio 5:1) and has a population prevalence of 1-2%. A genetic determinant to the susceptibility to GD is suspected because of familial clustering of the disease, a high sibling recurrence risk, and the familial occurrence of thyroid autoantibodies. GD is a systemic autoimmune thyroid disorder characterized by the infiltration of immune effector cells and thyroid-antigen-specific T cells into the thyroid and thyroid stimulating hormone receptor (TSHR) expressing tissues, i.e. orbit, skin, with the production of autoantibodies to well-defined thyroidal antigens. Stimulatory autoantibodies in GD activate the TSHR leading to thyroid hyperplasia and unregulated thyroid hormone production and secretion. Diagnosis of GD is straightforward in a patient with a diffusely enlarged, heterogeneous, hypervascular (increased Doppler flow on neck ultrasound) thyroid gland, associated orbitopathy, biochemically confirmed thyrotoxicosis, positive TSHR autoantibodies, and often a family history of autoimmune disorders.
Introduction and pathomechanism
Thyrotoxicosis is defined as the state of thyroid hormone excess and is synonymous with hyperthyroidism, which is the result of excessive thyroid function. The major etiologies of thyrotoxicosis are hyperthyroidism caused by Graves’ disease (GD), toxic multinodular goiter, and toxic adenomas. GD accounts for 60 to 80% of thyrotoxicosis, though the prevalence varies among populations, depending mainly on iodine intake[1]. GD occurs more often in women than in men with a female: male ratio of 5:1 and a population prevalence of 1-2%[2]. The disorder rarely begins before adolescence and typically occurs between 20 and 50 years of age, though it also occurs in the elderly[3].GD is an autoimmune thyroid disorder characterized by the infiltration of immune effector cells and thyroid-antigen- specific T cells into the thyroid and thyroid stimulating hormone receptor (TSHR) expressing tissues, with the production of autoantibodies to well-defined thyroidal antigens such as thyroid peroxidase, thyroglobulin, and the TSHR. A genetic determinant to the susceptibility to GD is suspected because of familial clustering of the disease[4],[5], a high sibling recurrence risk, the familial occurrence of thyroid autoantibodies and concurrent autoimmune diseases[6],[7], and the 30% concordance in disease status between identical twins[8],[9]. Smoking and other lifestyle factors also increase the risk for Graves ́ hyperthyroidism[10]. The TSHR expressed on the plasma membrane of thyroid epithelial cells, is central to the regulation of thyroid growth and function. However, it is also expressed on a variety of other tissues, including adipocytes and bone cells. The TSHR is the major autoantigen in the autoimmune hyperthyroidism of GD where T cells and autoantibodies are directed at the TSHR antigen. Stimulatory autoantibodies in GD activate TSHR on thyroid follicular cells, leading to thyroid hyperplasia and unregulated thyroid hormone production and secretion[11].
The close clinical relationship between Graves’ hyperthyroidism and Graves’ orbitopathy (GO) has suggested that immunoreactivity against TSHR present in both the thyroid and orbit underlies both conditions[12]. A prerequisite for involvement of TSHR as an autoantigen in GO is that it be expressed in affected orbital tissues[13]. Numerous studies did demonstrate that TSHR mRNA and protein are present in GO. Further, TSHR expression has been shown to be higher in GO orbital fat compared with normal orbital adipose tissues. Also, there exists a positive correlation between TSHR mRNA levels in individual GO orbital connective tissue specimens and the patient’s clinical disease activity[14]. The extrathyroidal manifestations of GD i.e. GO and dermopathy are due to immunologically mediated activation of fibroblasts in the extraocular muscles and skin, with accumulation of glycosaminoglycans, leading to the trapping of water and edema[15]. Later, fibrosis becomes prominent. The fibroblast activation is caused by proinflammatory cytokines derived from locally infiltrating T cells and macrophages[16].
Clinical Spectrum
Table 1:
Causes of Thyrotoxicosis
Primary hyperthyroidism
- Graves’ disease
- Toxic multinodular goiter
- Toxic adenoma
- Amiodarone, iodine excess
- Ingestion of excess thyroid hormone (thyrotoxicosis factitia) or thyroid tissue
- Subacute thyroiditis
- Silent thyroiditis
- Activating mutation of the TSH receptor (autosomal dominant)
- Struma ovarii
- Functioning thyroid carcinoma metastases
- TSH-secreting pituitary adenoma
- Thyroid hormone resistance syndrome
- Chorionic gonadotropin-secreting tumors
- Gestational thyrotoxicosis
Table 2:
Signs and Symptoms of Thyrotoxicosis
Symptoms
- Hyperactivity, irritability
- Heat intolerance and sweating
- Palpitations
- Dysphoria
- Fatigue and weakness
- Weight loss with increased appetite
- Diarrhea
- Polyuria
- Oligomenorrhea, loss of libido
- Tachycardia
- Atrial fibrillation in the elderly
- Tremor
- Goiter
- Warm, moist skin
- Muscle weakness, proximal myopathy
- Lid retraction or lag
- Gynecomastia
In GD the thyroid is usually diffusely enlarged to two to three times its normal size. The consistency is firm, but less so than in multinodular goiter. There may be a thrill or bruit due to the increased vascularity of the gland and the hyperdynamic circulation. The most common cardiovascular manifestation is sinus tachycardia, often associated with palpitations and sometimes due to supraventricular tachycardia. The high cardiac output produces a bounding pulse, widened pulse pressure, and an aortic systolic murmur, and can lead to worsening of angina or heart failure in the elderly or those with preexisting heart disease[19]. Atrial fibrillation is more common in patients > 50 years. Treatment of the thyrotoxic state alone reverts atrial fibrillation to normal sinus rhythm in fewer than half of patients, suggesting the existence of an underlying cardiac problem in the remainder.
The skin is usually warm and moist, and the patient complains of sweating and heat intolerance, particularly during warm weather. Palmar erythema, onycholysis, and less commonly, pruritus, urticaria, and diffuse hyperpigmentation may be evident. Hair texture may become fine, and a diffuse alopecia occurs in up to 40% of patients, persisting for months after restoration of euthyroidism.
Fine tremor is a very frequent finding, best elicited by asking patients to stretch out the fingers and feeling the fingertips with the palm. Common neurologic manifestations include hyperreflexia, muscle wasting, and proximal myopathy without fasciculation. Chorea is a rare feature. Thyrotoxicosis is sometimes associated with a form of hypokalemic periodic paralysis; this disorder is particularly common in Asian males with thyrotoxicosis. Gastrointestinal transit time is decreased, leading to increased stool frequency, often with diarrhea and occasionally mild steatorrhea. Women frequently experience oligomenorrhea or amenorrhea; in men there may be impaired sexual function and, rarely, gynecomastia.
The direct effect of thyroid hormones on bone resorption leads to osteopenia in long-standing thyrotoxicosis; mild hypercalcemia occurs in up to 20% of patients, but hypercalcuria is more common. There is a small increase in fracture rate in patients with a previous history of thyrotoxicosis.
Extrathyroidal manifestations
Lid retraction, causing a staring appearance, can occur in any form of thyrotoxicosis and is the result of sympathetic overactivity. However, GD is associated with specific eye signs that comprise Graves’ orbitopathy (GO). This condition may occur in the absence of GD in 10% of patients. Most of these individuals have autoimmune hypothyroidism or thyroid antibodies. The onset of GO occurs within the year before or after the diagnosis of thyrotoxicosis in 75% of patients but can sometimes precede or follow thyrotoxicosis by several years, accounting for some cases of euthyroid GO. Many patients with GD have little clinical evidence of GO. However, the enlarged extraocular muscles typical of the disease, and other subtle features, can be detected in almost all patients when investigated by ultrasound or computed tomography (CT) imaging of the orbits[20]. Unilateral signs are found in up to 10% of patients.The earliest manifestations of GO are a sensation of grittiness, eye discomfort, and excess tearing. About a third of patients have proptosis, best detected by visualization of the sclera between the lower border of the iris and the lower eyelid, with the eyes in the primary position. Proptosis can be measured using an exophthalmometer. In severe cases, proptosis may cause corneal exposure and damage, especially if the lids fail to close during sleep. Periorbital edema, scleral injection, and chemosis are also frequent. In 5 to 10% of patients, the muscle swelling is so severe that diplopia results, typically but not exclusively when the patient looks up and laterally. The most serious manifestation is compression of the optic nerve at the apex of the orbit, leading to papilledema, peripheral field defects, and, if left untreated, permanent loss of vision.
Clinical features of GO vary from a mild grittiness of the eyes to severe diplopia, disfiguring proptosis and loss of vision. There is a natural tendency towards spontaneous improvement: the spontaneous course depicts an active phase, which slowly abates after which an inactive phase ensues[21]. The most common symptoms of GO are eyelid retraction (90%), soft tissue involvement (80%), proptosis (50-60%), dry eye syndrome (50%), motility disorders (40%), optic neuropathy (3-5%) and superior limbic keratitis (2%)17. The autoimmune process leads to an accumulation of collagen and hydrophilic glycosaminoglycans within the orbit. Inflammatory changes of the eyelids cause visible edema and erythema. If extraocular muscles are affected motility disorders may occur. Patients with these motility disturbances, with diplopia, severe and active disease have a severely impaired health related quality of life[22].
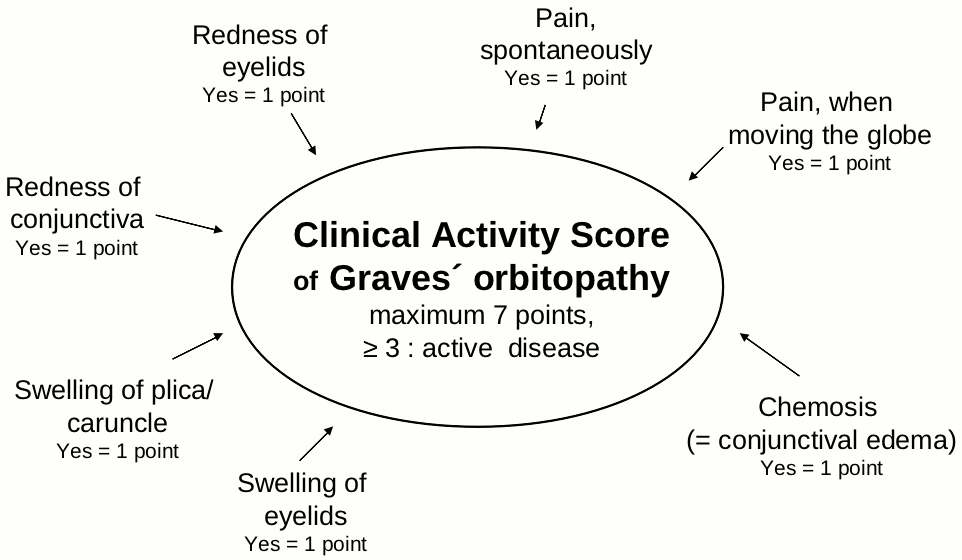
Activity and severity of GO
Many scoring systems have been used to gauge the extent and severity of the orbital changes in GD. The NOSPECS scheme[23],[24] includes six classes of eye changes (Table 3). GO is classified as severe if corneal involvement, severe proptosis, constant diplopia or optic neuropathy are present[25].Table 3:
The Clinical Severity Score according to the “NOSPECS classification”, modified according to [23],[24].| NOSPECS-Class | 0 | I | II (soft tissue involvement) |
III (proptosis) |
IV (extraocular muscle involvement) |
V (corneal involvement) |
VI (sight loss due to optic nerve compression) |
|---|---|---|---|---|---|---|---|
| Grade | No physical signs or symptoms | Only signs | 0 = absent | 0 = absent | 0 = absent | 0 = absent | 0 = absent |
| a = minimal | a = minimal | a = limitation of motion in extremes of gaze | a = stipping of the cornea | a = minimal | |||
| b = moderate | b = moderate | b = evident restriction in motion | b = ulceration | b = moderate | |||
| c = marked | c = marked | c = fixation of a globe | c = necrosis, perforation | c = marked (no light perception) |
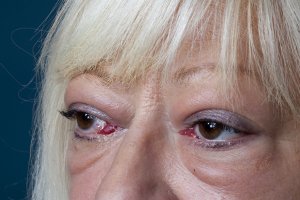
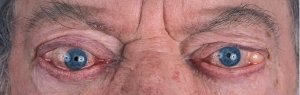
General ophthalmic assessment should include examination of anterior and posterior eye segment, applanation tonometry, Hertel exophthalmometry, and motility tests. Additionally, the observer classifies whether there are optic disc oedema or disc pallor and records whether choroidal folds are present. In addition to fundoscopy relative afferent pupillary defects, vision field defects, colour vision abnormalities, visual evoked potentials and visual acuity are tested in order to exclude optic neuropathy.
Cigarette smoking can profoundly influence the occurrence and the course of Graves’ eye disease[28], and also impairs its response to conservative treatment[29]. Accordingly, patients should be strongly urged to stop smoking. Refraining from smoking favorably influences the course of GO. Also, emotional distress and stressful life events are risk factors for GO and should therefore be minimized[30], [31].
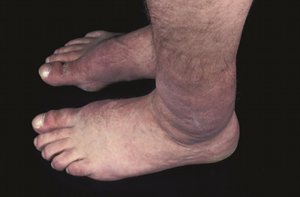
Graves’ dermopathy and Graves’ acropachy
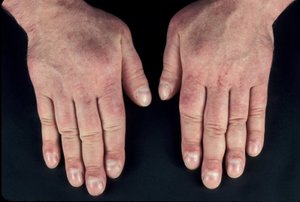
Laboratory evaluation and imaging
In GD, below-normal to suppressed levels of baseline serum TSH, normal to elevated serum levels of T4, elevated serum levels of T3 and of TSHR autoantibodies, as well as a diffusely enlarged, heterogeneous, hypervascular thyroid gland (increased Doppler flow in the ultrasound evaluation of the neck) confirm diagnosis of GD. In 2 to 5% of patients (and more in areas of borderline iodine intake), only T3 is increased (T3 toxicosis). The converse state of T4 toxicosis, with elevated total and free T4 and normal T3 levels, is occasionally seen when hyperthyroidism is induced by excess iodine, providing surplus substrate for thyroid hormone synthesis. Associated abnormalities that may cause diagnostic confusion in thyrotoxicosis include elevation of bilirubin, liver enzymes, and ferritin. Microcytic anemia and thrombocytopenia occur less often.
The clinical relevance of anti-TSHR antibodies
During entire pregnancy of patients with GD circulating anti-TSHR-autoantibodies can pass to the baby and cause either neonatal autoimmune thyrotoxicosis (functionally stimulating immunoglobulins) or hypothyroidsm (blocking autoantibodies). Currently, two different methods of assessing antibodies directed against TSHR are used. The TSHR binding inhibitory immunoglobulin (TBII) assay detects immunoglobulins that inhibit the binding of TSH to purified or recombinant TSHR. It thus measures both thyroid-stimulating (TSI) and thyroid-blocking antibodies that target the receptor. The second method is a bioassay that can distinguish between TSHR-stimulating, -neutral (binding) and –blocking autoantibodies through their effect on cyclic adenosine monophosphate (cAMP) production in a cell line stably transfected with the receptor[27],[35],[36],[37]. The levels of TSI closely correlate with activity and severity of GO[33], and in approximately 50% of the cases also are of prognostic value regarding the course of the disease[38].The commercially available TBII tests that are used to measure the binding of sera to TSHR display high sensitivity and specificity for TSHR autoantibodies, but unfortunately do not measure the functional activity of immunoglobulins and do not distinguish between stimulatory, blocking or neutral activity. In contrast, anti-TSHR bioassays offer the following advantages: 1) the biological activity of specific immunoglobulins is directly assessed on a fully functional TSHR holoreceptor expressed on intact live cells, a platform that is easily adaptable and tailored to detect antibodies of specific function; 2) the bioassay measures the specific function of autoantibody that highly correlates with Graves’ activity; 3) the monitoring of TSI levels and TSI titers add another dimension to the assessment of GO severity in individual patients.
Differential Diagnosis
Diagnosis of GD is straightforward in a patient with biochemically confirmed thyrotoxicosis, diffuse goiter on palpation, associated GO, positive TSHR antibodies, and often a personal or family history of autoimmune disorders[1],[2]. For patients with thyrotoxicosis who lack these features, the most reliable diagnostic methods are ultrasound evaluation of the neck looking for a hypervascular gland (“thyroid storm”) and/or a radionuclide scan of the thyroid, which will distinguish the diffuse, high uptake of Graves’ disease from nodular thyroid disease, destructive thyroiditis, ectopic thyroid tissue, and factitious thyrotoxicosis. In secondary hyperthyroidism due to a TSH-secreting pituitary tumor, there is also a diffuse goiter. The presence of a non suppressed TSH level and the finding of a pituitary tumor on CT or magnetic resonance imaging (MRI) scan readily identify such patients[20]. MRI is the optimal imaging procedure for the differential diagnosis of GO[39]. Clinical features of thyrotoxicosis can mimic certain aspects of other disorders including panic attacks, mania, pheochromocytoma, and the weight loss associated with malignancy. The diagnosis of thyrotoxicosis can be easily excluded if the TSH level is normal. A normal TSH also excludes GD as a cause of diffuse goiter.
Clinical Course of Graves’ disease
Clinical features generally worsen without treatment; mortality was 10 to 30% before the introduction of satisfactory therapy. Some patients with mild GD experience spontaneous relapses and remissions. Rarely, there may be fluctuation between hypothyroidism and hyperthyroidism due to changes in the functional activity of TSHR antibodies. About 15% of patients who enter remission after conservative treatment develop hypothyroidism 10 to 15 years later as a result of the destructive autoimmune process. The clinical course of GO does not follow that of the thyroid disease. GO typically worsens over the initial 3 to 6 months, followed by a plateau phase over the next 12 to 18 months, with spontaneous improvement, particularly in the soft tissue changes. However, the course is more fulminant in up to 5% of patients, requiring intervention in the acute phase if there is optic nerve compression or corneal ulceration. Diplopia may appear late in the disease due to fibrosis of the extraocular muscles. Radioiodine treatment for hyperthyroidism worsens the eye disease[40] in approximately 15-20% of patients (especially smokers). Antithyroid drugs or surgery have no adverse effects on the clinical course of GO[41]. Dermopathy, when it occurs, usually appears 1 to 2 years after the development of Graves’ hyperthyroidism; it may improve spontaneously.- Brent GA. Clinical practice. Graves' disease. N Engl J Med 2008 Jun 12; 358(24): 2594-605.
- Vanderpump MP, Tunbridge WM, French GM. The incidence of thyroid disorders in the community; a twenty year follow-up. Clin Endocrinol 1995; 43: 55-68
- Pearce SHS. Graves’ disease. In Weetman AP (Ed) Autoimmune diseases in endocrinology. Humana press, Totowa, NJ, USA, pp 117-135
- Dittmar M, Libich C, Brenzel T, Kahaly GJ. Increased Familial Clustering of Autoimmune Thyroid Diseases. Horm Metab Res. 2011 Feb 1. [Epub ahead of print]
- Dultz G, Matheis N, Dittmar M, Bender K, Kahaly GJ. CTLA-4 CT60 polymorphism in thyroid and polyglandular autoimmunity. Horm Metab Res. 2009 Jun;41(6):426-9.
- Teufel A, Weinmann A, Kahaly GJ, Centner C, Piendl A, Wörns M, Lohse AW, Galle PR, Kanzler S. Concurrent autoimmune diseases in patients with autoimmune hepatitis. Clin Gastroenterol. 2010 Mar;44(3):208-13.
- Boelaert K, Newby PR, Simmonds MJ, Holder RL, Carr-Smith JD, Heward JM, Manji N, Allahabadia A, Armitage M, Chatterjee KV, Lazarus JH, Pearce SH, Vaidya B, Gough SC, Franklyn JA. Prevalence and relative risk of other autoimmune diseases in subjects with autoimmune thyroid disease. Am J Med. 2010 Feb;123(2):183.e1-9.
- Vaidya B, Kendall-Taylor P, Pearce SHS. The genetics of autoimmune thyroid disease. J Clin Endocrinol Metab 2002; 87: 5385-5397
- Brix TH, Kyvik KO, Christensen K, Hegedus L. Evidence for a major role of heredity in Graves’ disease: a population-based study of two Danish twin cohorts. J Clin Endocrinol Metab 2001; 86: 930-934
- Holm IA, Manson JE, Michels KB, Alexander EK, Willet WC, Utiger RD. Smoking and other lifestyle factors and the risk of Graves‘ hyperthyroidism. Arch Int Med 2005; 165: 1606-1611
- Weetman AP. Determinants of autoimmune thyroid disease. Nat Immunol 2001; 2: 769-770
- Lazarus J, Marino M. Orbit-thyroid relationship. In Wiersinga WM, Kahaly GJ (Editors): Graves’ orbitopathy – A multidisciplinary approach – Questions and answers, 2nd revised edition, Karger, Basel, pp 26-32
- Kahaly GJ. The thyrocyte-fibrocyte link: closing the loop in the pathogenesis of Graves' disease? J Clin Endocrinol Metab. 2010 Jan;95(1):62-5
- Bahn RS. Graves’ opthalmopathy. N Engl J Med. 2010 Feb 25;362(8):726-38
- Orgiazzi J, Ludgate M. Pathogenesis. In Wiersinga WM, Kahaly GJ (Editors): Graves’ orbitopathy – A multidisciplinary approach – Questions and answers, 2nd revised edition, Karger, Basel, pp 40-56
- Kahaly GJ, Shimony O, Gellman YN, Lytton SD, Eshkar-Sebban L, Rosenblum N, Refaeli E, Kassem S, Ilany J, Naor D. Regulatory T-cells in graves' orbitopathy: baseline findings and immunomodulation by anti-T lymphocyte globulin. J Clin Endocrinol Metab. 2011 Feb;96(2):422-9
- Bartalena L, Tanda ML. Clinical practice. Graves' ophthalmopathy. N Engl J Med. 2009 Mar 5;360(10):994-1001.
- Boelaert K, Torlinska B, Holder RL, Franklyn JA. Older subjects with hyperthyroidism present with a paucity of symptoms and signs: a large cross-sectional study. J Clin Endocrinol Metab. 2010 Jun;95(6):2715-26.
- Biondi B, Kahaly GJ. Cardiovascular involvement in patients with different causes of hyperthyroidism. Nat Rev Endocrinol. 2010 Aug;6(8):431-43.
- Kahaly GJ. Imaging in thyroid-associated orbitopathy. Eur J Endocrinol. 2001 Aug;145(2):107-18.
- Hales IB, Rundle FF. Ocular changes in Graves' disease. A long-term follow-up study. Q J Med. 1960 Jan;29:113-26
- Ponto KA, Kahaly GJ. Quality of life in patients suffering from thyroid orbitopathy. Pediatr Endocrinol Rev. 2010 Mar;7 Suppl 2:245-9
- Werner SC. Modification of the classification of the eye changes in Graves´ disease. Am J Ophthalmol 1977, 83(5):725-727
- Dickinson AJ. Clinical Manifestation. In: Wiersinga WM and Kahaly GJ (Editors): Graves’ Orbitopathy - A multidisciplinary Approach - Questions and Answers, 2nd, revised Edition, Karger, Basel, 2010, 1-25
- Bartalena L, Baldeschi L, Dickinson AJ, Eckstein A, Kendall-Taylor P, Marcocci C, Mourits MP, Perros P, Boboridis K, Boschi A, Currò N, Daumerie C, Kahaly GJ, Krassas G, Lane CM, Lazarus JH, Marinò M, Nardi M, Neoh C, Orgiazzi J, Pearce S, Pinchera A, Pitz S, Salvi M, Sivelli P, Stahl M, von Arx G, Wiersinga WM. Consensus statement of the European group on Graves' orbitopathy (EUGOGO) on management of Graves' orbitopathy. Thyroid. 2008 Mar;18(3):333-46
- Mourits MP, Koornneef L, Wiersinga WM, Prummel MF, Berghout A, van der Gaag R. Clinical criteria for the assessment of disease activity in Graves' ophthalmopathy: a novel approach. Br J Ophthalmol. 1989 Aug;73(8):639-44
- Lytton SD, Ponto KA, Kanitz M, Matheis N, Kohn LD, Kahaly GJ. A novel thyroid stimulating immunoglobulin bioassay is a functional indicator of activity and severity of Graves' orbitopathy. J Clin Endocrinol Metab. 2010 May;95(5):2123-31
- Bartalena L, Martino E, Marcocci C, Bogazzi F, Panicucci M, Velluzzi F, Loviselli A, Pinchera A. More on smoking habits and Graves' ophthalmopathy. J Endocrinol Invest 1989 Nov;12(10):733-7
- Bartalena L, Marcocci C, Tanda ML, Manetti L, Dell'Unto E, Bartolomei MP, Nardi M, Martino E, Pinchera A. Cigarette smoking and treatment outcomes in Graves’ ophthalmopathy. Ann Intern Med 1998: 15;129(8):632-5
- Winsa B, Adami HO, Bergström R, Gamstedt A, Dahlberg PA, Adamson U, Jansson R, Karlsson A. Stressful life events and Graves' disease. Lancet. 1991 Dec 14;338(8781):1475-9
- Yoshiuchi K, Kumano H, Nomura S, Yoshimura H, Ito K, Kanaji Y, Kuboki T, Suematsu H. Psychosocial factors influencing the short-term outcome of antithyroid drug therapy in Graves' disease. Psychosom Med. 1998 Sep-Oct; 60(5):592-6
- Fatourechi V, Bartley GB, Eghbali-Fatourechi GZ, Powell CC, Ahmed DD, Garrity JA. Graves' dermopathy and acropachy are markers of severe Graves' ophthalmopathy. Thyroid 2003 Dec;13(12):1141-4
- Gerding MN, van der Meer JW, Broenink M, Bakker O, Wiersinga WM, Prummel MF. Association of thyrotrophin receptor antibodies with the clinical features of Graves' ophthalmopathy Clin Endocrinol (Oxf) 2000; 52(3):267-71
- Schwartz KM, Fatourechi V, Ahmed DD, Pond GR. Dermopathy of Graves' disease (pretibial myxedema): long-term outcome. J Clin Endocrinol Metab 2002 Feb;87(2):438-46
- Lytton SD, Li Y, Olivo PD, Kohn LD, Kahaly GJ. Novel chimeric thyroid-stimulating hormone-receptor bioassay for thyroid-stimulating immunoglobulins. Clin Exp Immunol. 2010 Dec;162(3):438-46
- Kamijo K, Murayama H, Uzu T, Togashi K, Kahaly GJ. A novel bioreporter assay for thyrotropin receptor antibodies using a chimeric thyrotropin receptor (mc4) is more useful in differentiation of Graves' disease from painless thyroiditis than conventional thyrotropin-stimulating antibody assay using porcine thyroid cells. Thyroid. 2010 Aug;20(8):851-6.
- Lytton SD, Kahaly GJ. Bioassays for TSH-receptor autoantibodies: an update. Autoimmun Rev. 2010 Dec;10(2):116-22.
- Eckstein AK, Plicht M, Lax H, Neuhäuser M, Mann K, Lederbogen S, Heckmann C, Esser J, Morgenthaler NG. Thyrotropin receptor autoantibodies are independent risk factors for Graves' ophthalmopathy and help to predict severity and outcome of the disease. J Clin Endocrinol Metab. 2006 Sep;91(9):3464-70
- Mourits MP. Diagmosis and Differential Diagnosis of Graves´ Orbitopathy. In Wiersinga WM, Kahaly GJ (Editors): Graves’ orbitopathy – A multidisciplinary approach – Questions and answers, 2nd revised edition, Karger, Basel, pp 66.76
- Ponto KA, Zang S, Kahaly GJ. The tale of radioiodine and Graves' orbitopathy. Thyroid. 2010 Jul;20(7):785-93.
- Marcocci C, Pinchera A. Thyroid treatment. In Wiersinga WM, Kahaly GJ (Editors): Graves’ orbitopathy – A multidisciplinary approach – Questions and answers, 2nd revised edition, Karger, Basel, pp 100-110